[Heavy Review] Cytoplasmic DNA and Aging and Diseases
The following article comes from the old urchin, and the author said that the old urchin
The old urchin said.
WeChat official account is committed to spreading the cutting-edge scientific research progress and interesting popular science related to aging, helping everyone to understand the scientific story behind aging more deeply ~

Follow us for more information.
Translated by Alva Wang, Xiong Muzhao, Cai Linguo and Yang Ping.
Novel coronavirus can cause immune response after being infected with human body. However, in addition to being infected with pathogenic microorganisms, human body can also produce immune response without being infected, that is, aseptic inflammation. Aseptic inflammation is an important factor leading to chronic diseases and individual aging. Recently, it has been found that there is an obvious correlation between cytoplasmic DNA and the activation pathway of aseptic inflammation. On October 28th, 2021, Prof. Peter D. Adams from Sanford burnham Presby Institute of Medicine and Prof. João F. Passos from Mayo Medical Center published a review entitled "Cytoplasmic DNA: Sources, Sensing, and Role in Aging and Disease" on the Cell, focusing on the formation of cytoplasmic DNA and the potential molecular mechanism of its related functions, providing new ideas for finding strategies to treat chronic diseases.
Abstract
Endogenous cytoplasmic DNA (CytoDNA) has been proved to be a key inflammatory mediator in many physiological and pathological processes. Although the role of cytoplasmic DNA in innate immune activation has been clarified, cytoplasmic DNA itself is often lacking in characteristics and difficult to be recognized, and its formation mechanism, functional scope and its role in diseases are not completely clear. In this paper, the author summarizes the research progress in this rapidly developing field, focusing on the similarities and differences between different cytoplasmic DNA, the potential molecular mechanism of their formation and function, the interaction between cytoplasmic DNA pathways, and the potential application in the treatment of aging-related diseases.
Jieshao
As the main medium for storing biological information, DNA is distributed in specific compartments of eukaryotic cells and separated from other cellular components. This kind of compartment, which exists in the nucleus or endosymbiont organelles (i.e. mitochondria and chloroplasts), can specialize and compartmentalize the main DNA functions and cell processes, thus regulating gene expression, genome replication and the repair of damaged DNA. In addition, cellular responses have evolved the functions of sensing, destroying and transmitting signals of exogenous DNA, and these functions are partly realized according to the information that they have located in cells that usually do not contain DNA. In the cells of multicellular animals, the above cellular reactions are part of the innate immunity of organisms and can be activated by various stimuli (such as virus and bacterial infections).
A similar pathway may also be activated by misplaced endogenous cytoplasmic DNA, thus driving the cell’s autonomous response without infection. Endogenous cellular DNA is a trigger of aseptic inflammation. The so-called aseptic inflammation is inflammation without pathogenic infection, which is related to the occurrence and development of many chronic diseases, including cancer, cardiovascular diseases and neurodegenerative diseases. The change of immune function, such as aseptic inflammation, is considered as an important sign of aging process, also known as "inflammatory aging". Although the interaction between viral DNA and host defense has been thoroughly studied, the role of cytoplasmic DNA in aging and chronic diseases has not been recognized until recently. In this review, the author focuses on four kinds of endogenous cellular DNA: micronucleus, cytoplasmic chromatin fragment, mitochondrial DNA and retrotransposon, and also evaluates the main types of endogenous cellular DNA, clarifying the mechanisms related to the formation, function and pathology of cytoplasmic DNA.
Micronucleus
Micronucleus (Mn) is a complete chromosome or chromosome fragment wrapped by nuclear membrane in cytoplasm, which may be directly separated from the nucleus, or is usually thought to be caused by mitotic defects.
Formation of micronucleus
The formation of micronucleus occurs under the background of cell division, which is related to two related mechanisms of chromosome error division, including: (1) Mitotic defects include metaphase or defective metacentric-kinetochore assembly or anaphase abnormality, and (2)DNA repair abnormality (Figure 1A). Although mitotic defects are related to the wrong separation of the whole "lagging" chromosome in the later stage, abnormal DNA repair is related to the wrong separation of chromosome fragments. These fragments can be formed by the fusion of chromosome fragments caused by unrepaired DNA double strand breaks (DSBs) or the wrong repair of telomere ends of DSBs. The former can lead to the wrong segregation of chromosome fragments lacking centromere, while the latter can lead to the formation of chromosome bridges. The fusion chromosome containing two centromeres breaks during cytokinesis, or can persist in the later stage of mitosis, and forms a bridge structure between the two newly formed nuclei. The breakage of this bridge leads to the extensive rearrangement of the bridge region through non-homologous terminal connection, and the formation of chromosome fragments lacking centromere or with other centromere defects. These abnormal chromosomes can produce micronucleus in the next round of cell division. In this case, the wrongly separated nuclear DNA will form a single nuclear membrane and become micronucleus, which will affect the cell function and fate.
Function of micronucleus
Micronucleus is initially surrounded by nuclear membrane, which separates micronucleus from cytoplasm. However, the integrity of the nuclear membrane may be lost spontaneously, leading to rupture (Figure 1B). Micronuclei carry defective nuclear membrane components, which are characterized by the lack of nuclear pore complexes and the weakening of nuclear transport capacity, which will affect many biological processes in micronucleus, such as the defects of replication and repair of micronucleus DNA. The loss of nuclear membrane integrity of micronucleus is also related to the rupture of micronucleus and the induction of micronucleus DNA by cell DNA receptors.
Although many DNA receptors have been reported, the most thoroughly studied cytoplasmic DNA receptor is cyclic GMP-AMP synthetase (cGAS). After recognizing DNA, the activated cGAS uses ATP and GTP as substrates to generate the second messenger loop GMP-AMP(cGAMP), which will bind and activate IFN gene stimulator (STING). STING oligomerization induced by cGAMP provides a signal platform for the recruitment and activation of TANK-binding kinase (TBK1). STING can also be used as a scaffold for phosphorylation of IRF3 by TBK1 and subsequent dimerization of IRF3. The dimerized IRF3 will enter the nucleus and trigger the type I interferon response. STING-related TBK1 also promotes the activation of nuclear factor κB(NF-κB) mediated by dsDNA to promote the transcription of inflammation-related genes. Gene knockout studies in mice show that cGAS is necessary for DNA ligand and virus-induced interferon response, which indicates that cGAS is the main cytoplasmic DNA receptor in these cases. Similarly, cGAS activation due to the rupture of micronucleus nuclear membrane is considered to be the main trigger of interferon-based inflammatory response.
However, the exact relationship between micronucleus rupture, DNA damage and cGAS sensing is still unclear. In view of the fact that some cGAS are usually located in nucleosomes, and the activation of cGAS by DNA/ chromatin binding is highly regulated (see prospect and conclusion for details), micronucleus breakage alone is not enough to activate cGAS. Similar to nuclear rupture, micronucleus rupture is related to extensive DNA damage, which may be due to the exposure of micronucleus to cytoplasmic factors, such as exonuclease TREX1 or other nucleases, so DNA damage may directly or indirectly promote cGAS activation. The perception of micronucleus by cGAS plays an environmentally dependent role in inflammatory signals and tumorigenesis. Mackenzie and others speculate that cGAS recognition of micronucleus DNA may be used as an intracellular immune monitoring mechanism to monitor a series of tumor induction processes (that is, micronucleus induction is a tumor inhibition mechanism). Harding et al.’ s research also suggests that cGAS/STING’s perception of micronucleus has tumor inhibitory effect, which may be mainly in the early stage of disease progression. Therefore, cancer cells usually inactivate cGAS-STING pathway to avoid immune monitoring.
Dysfunction of micronucleus
Although micronucleus is thought to be activated as a tumor inhibition pathway, it also drives genome instability through chromosome fragmentation, and plays a pathogenic role in the evolution and metastasis of cancer genome. Chromosome fragmentation is a widespread process of chromosome breakage, and some of it is considered to occur through the cycle of broken fusion bridges and the accumulation of DNA damage in micronucleus. Compared with micronucleus, the nucleoplasmic bridge formed between daughter nuclei has similar defects in nuclear membrane assembly, which leads to replication and repair defects and eventually rupture. This breakage and subsequent cell division can start the cycle of micronucleus formation, damage and reintegration into the genome through non-homologous terminal connection, further aggravating the genomic instability of chromosome fragmentation and evolution. In addition, the abnormal activation of STING-dependent NF-κB signal will also promote tumor metastasis due to micronucleus rupture.
Therapeutic target of micronucleus
The micronucleus activation of cGAS-STING signal is the internal mechanism of monitoring genomic instability, which is of great significance for cancer treatment. Therefore, activating the signal pathway downstream of micronucleus, especially STING, is a potential target in cancer immunotherapy. However, we need to pay attention, because in some cases, the activation of cGAS-STING pathway or other candidate micronucleus receptors is tumor-promoting. For example, in breast cancer and lung cancer, inhibiting chromosome instability and related cytoplasmic DNA can reduce the metastasis of cancer cells. This may be related to the environment-dependent utilization of cGAS-STING downstream pathway in changing immune function, the participation of other cell DNA receptors, the role of tissue environment in different environments or some combinations of these factors. In the case of chronic STING activation in aging cells near the tumor, STING activation may also promote tumor occurrence, which suggests that other environments of cytoplasmic DNA biology in non-tumor cells must also be considered for drug targeting of STING and cGAS.
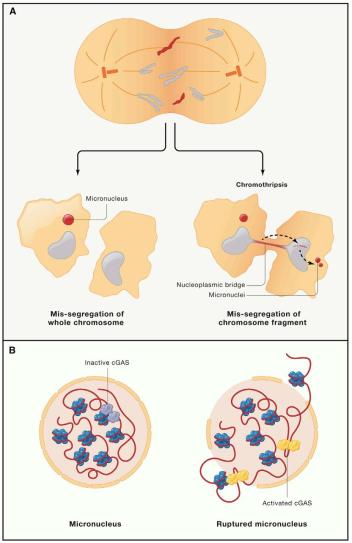
Fig. 1 Micronucleus formation mechanism
(a) Micronucleus is caused by chromosome segregation defect, which is caused by chromosome segregation error or DNA repair error during mitosis. The formation of abnormal chromosomes may directly lead to the formation of micronucleus or nuclear bridge. The fracture and reassembly of nuclear bridge can form micronucleus in the next round of cell division. (b) Micronucleus is characterized by nuclear membrane instability, and nuclear membrane rupture leads to cGAS activation.
Cytoplasmic chromatin fragment
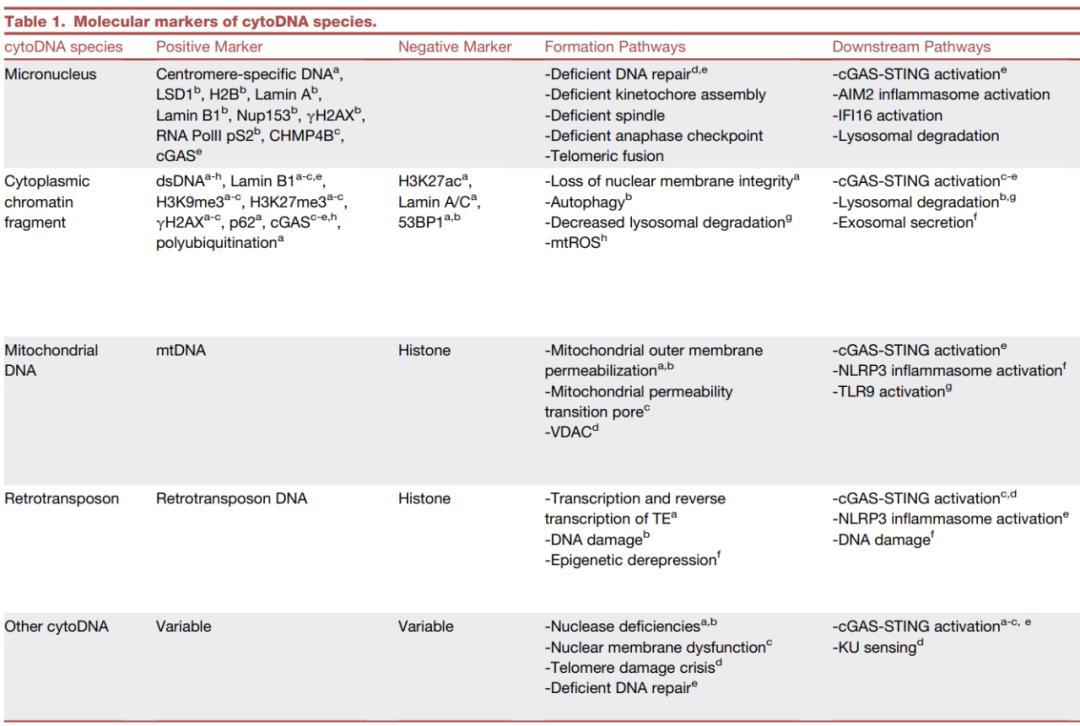
Cell senescence is one of the inducing factors of age-related aseptic inflammation and diseases, which is a stress response of cells to severe stimuli, mainly manifested in morphological and metabolic changes, permanent exit from the cell cycle, cell death arrest, and the formation of inflammatory aging-related secretory phenotype (SASP). There are chromatized micronucleus-like structures in the cytoplasm of aging cells. However, unlike real micronucleus. Many of these structures are formed independently of the cell cycle process. Its formation mode is that chromatin bubbles and enters the cytoplasm from the nucleus, and contains different molecular markers (Table 1), which indicates that the formation mechanism of these cytoplasmic chromatin fragments (CCF) is different from the real micronucleus.
Formation of CCF
CCF is characterized by a series of chromatin modifications, including the existence of heterochromatin-related H3K27me3 and the deletion of euchromatin-related H3K9Ac. This shows that CCF is formed by heterochromatin, although the genomic sites where these changes occur are not clear. The formation of CCF is related to the loss of nuclear membrane integrity in aging. To some extent, the integrity of the nuclear membrane depends on the nuclear fiber layer (a network structure composed of fibrin between A, B1, B2 and C, which supports the nuclear membrane). The loss of lamin B1 is also a recognized feature of cell senescence. The formation of CCF is also related to autophagy, which is a series of life activities to maintain intracellular homeostasis and is used to transport and degrade macromolecules and cellular components. The formation of CCF is also related to the autophagy degradation of Lamin B1 in aging cells. However, it is not clear whether the down-regulation of Lamin B1 is the cause or consequence of CCF formation. It is known that the knock-out of Lamin B1 can promote the formation of CCF-like lesions, and the over-expression of Lamin B1 can delay the aging process, which indicates that the loss of Lamin B1 may be the upstream regulatory factor of CCF formation. However, the formation of CCF in aging fibroblasts requires the interaction between Lamin B1 and LC3B (an autophagic substrate transfer protein) in the nucleus, and then Lamin B1 containing CCF in the cytoplasm undergoes autophagy degradation. This shows to some extent that,CCF is the carrier for degradation of Lamin B1. In addition, there is evidence that the mRNA level of Lamin B1 in aging cells is decreased, which indicates that there are many mechanisms to regulate the expression of Lamin B1 during aging. Clarifying the kinetics of Lamin B1 expression and degradation in aging cells will help to explain the relationship between CCF formation and autophagy.
CCF is strongly positive for DNA damage marker γH2AX, but it lacks a typical negative regulator of CCF formation, namely co-located DNA repair protein 53BP1. The observation that CCF is strongly positive for γH2AX shows that DNA damage plays an important role in the formation of CCF. DNA damage, especially the formation of DSB, is a powerful inducer of cell aging. Part of DNA damage is related to the change of mitochondrial function in aging cells through positive feedback loop and the continuous DNA damage caused by nuclear-mitochondrial (anterograde) and mitochondrial-nuclear (retrograde) pathways. Compared with cells in normal proliferation period or quiescent period, the mitochondrial mass of aging cells increases, the mitochondrial membrane potential decreases, the mitochondrial phagocytosis function may change, and the mitochondrial reactive oxygen species level increases. Interestingly, removing mitochondria from aging cells or inhibiting the formation of mtROS will inhibit SASP and prevent the formation of CCF, which indicates that mitotic nuclear pathway plays a role in the formation of CCF. It is reported that mtROS can activate stress-activated kinase JNK1/2 interacting with CCF inhibitor 53BP1 in mechanism. 53BP1 is a scaffold protein related to DNA damage, which recruits DNA repair factors into the DSB of DNA and inhibits the excision of the DSB end, which indicates that the formation of CCF requires the excision of the DSB end of DNA, and the inhibition of MRE11 by mirin also prevents the formation of CCF (Figure 2).Recently, similar mechanisms have been found in the genome instability models of human brain and Drosophila.
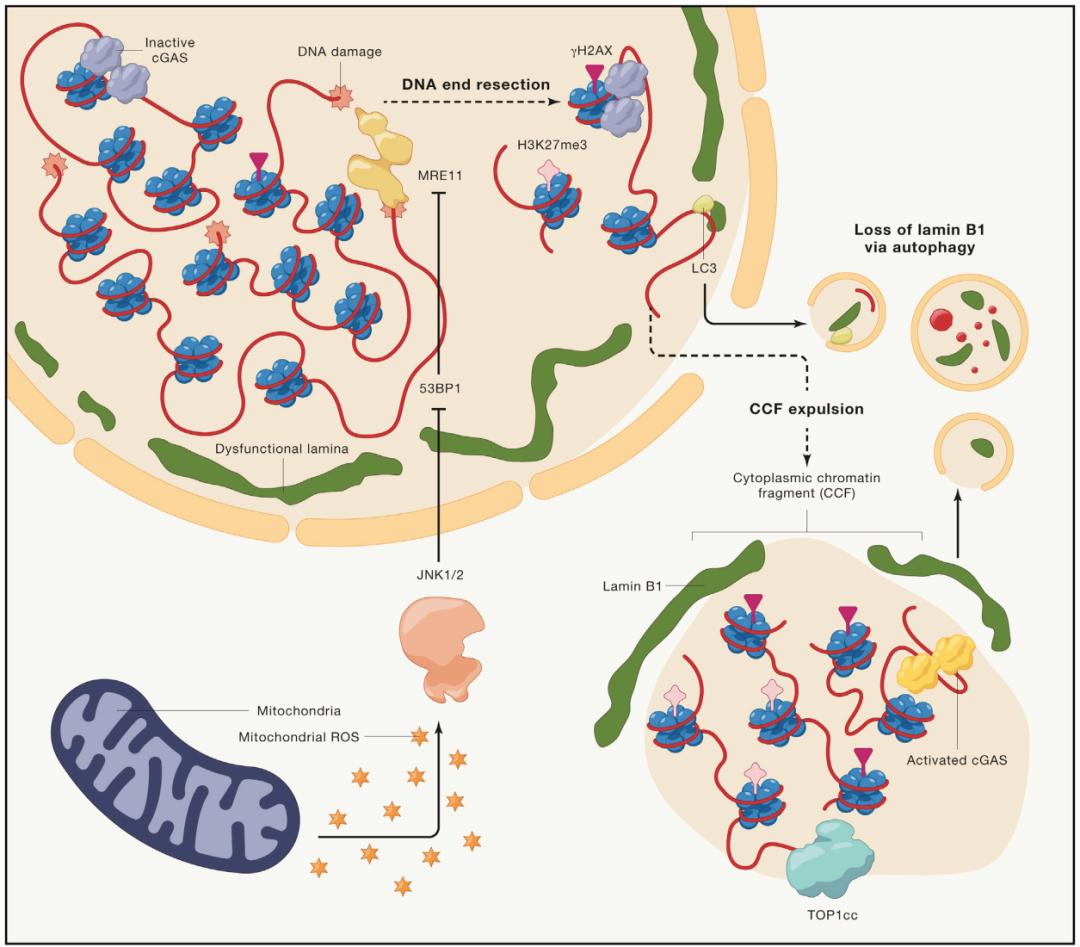
Fig. 2 Mechanism of CCF formation
The loss of Lamin B1 leads to the loss of nuclear membrane integrity through autophagy or other ways, and promotes the formation of CCF. Mitochondria can activate JNK1/2 signal pathway by producing mtROS, and participate in the formation of CCF. In the nucleus, 53BP1, as an inhibitor of CCF formation, may play a role by inhibiting MRE11-dependent double-stranded DNA cleavage. The markers of CCF include DNA damage marker γH2AX, heterochromatin marker H3K27me3, topoisomerase 1 cleavage complex (Top1Cc, which can enhance the binding of DNA to cGAS) and Lamin B1.
Functions of CCF
CCF will be induced by cGAS-STING pathway after entering cytoplasm (Figure 2). However, whether the DNA in CCF is a good substrate for cGAS activation and whether it can be obtained by cGAS remains to be studied. The activation of cGAS-STING leads to the activation of NF-κB of SASP gene downstream of STING, but not interferon gene, which may be due to the inhibition of interferon gene by p38 mitogen-activated protein kinase (MAPK) in aging cells. Under the condition of acute stress, aging cells, as an effective tumor inhibition mechanism, prevent cell growth and promote the elimination of potential malignant cells through SASP-dependent immune system activation. By observing the autophagic degradation of Lamin B1 in CCF, it is found that CCF may be the "reservoir" of nuclear autophagy substrate. At present, many other autophagy substrates have been found in aging cells. However, the direct role of CCF in this process is still unclear. In fact, CCF contains many chromatin-related proteins, but their fate in cytoplasm, such as autophagy degradation, independent signal transduction of cGAS and even cell secretion, is rarely reported. The correlation between γH2AX and CCF indicates that CCF is a by-product of DNA repair process. Most DNA repair factors are down-regulated in aging cells, which leads to the decrease of repair activity, especially the decrease of homologous repair pathway activity. This shows that,Removal and degradation of failed and potentially unstable repair intermediates (such as CCF) may be a mechanism to protect genome integrity or inhibit excessive DNA damage response, DDR) signal.
Table 1 molecular markers of cytoplasmic DNA
CCF and disease treatment
In addition to the functions of CCF and SASP mentioned above, these signals may also have harmful effects on human health. In some cases (such as aging), the failure to clear aging cells will lead to their accumulation and chronic SASP inflammation. A large number of studies show that aging cell accumulation and chronic SASP are the main factors leading to aging and related diseases. Therefore, the removal of aging cells by genetic or pharmacological "anti-aging" methods can prolong the life span of aged mice and prevent the occurrence of aging-related diseases. However, taking some anti-aging drugs for a long time may "accidentally injure" non-aging cells and produce toxic and side effects. As an alternative method, reducing SASP by "pathogenic" or "pathogenic" method may have lower toxic and side effects, and many studies have suggested that this method is effective in animal models.
Targeting the formation of CCF through the downstream targets of JNK signaling pathway or cGAS-STING pathway inhibitors can provide more strategies for the development of anti-aging drugs. Recent studies have shown that histone deacetylase inhibitors (HDACiS, such as voronostatin, an HDA CI approved for the treatment of some cancers in humans) can indirectly inhibit the formation of CCF and SASP by activating the mitochondrial function in aging cells and mice’s liver, which indicates that this method can play a targeted role in the treatment of aging in vivo. Further research on related mechanisms can find more targets (such as JNK1/2 and MRE11) to reduce human SASP, chronic inflammation and related chronic diseases. In view of the role of aging cells in promoting aging-related diseases, it is of great significance to analyze the interaction between mitochondrial function, autophagy and various cytoplasmic DNA during aging and the therapeutic potential of regulating these interactions (see prospect and conclusion for details).
Other forms of cytoplasmic DNA related to DNA damage
In addition to micronucleus in splinter cell and CCF formation in aging cells, there are several other cytoplasmic DNA labeled as micronucleus or CCF with unclear formation mechanism. These include cells from ataxia telangiectasia patients and cytoplasmic DNA; in Dnase2a deficient mouse model; Cytoplasmic DNA of CCF-like structure mediated by telomere damage, nuclear abnormality related to HGPS disease and cytoplasmic DNA in chromosome instability caused by excessive excision of Exo1; And small cytoplasmic DNA fragments related to DNA damage. The formation of these cytoplasmic DNA is directly or indirectly related to DNA damage marker γH2AX. However, it is not clear how these formation mechanisms are related to the specific mechanisms of micronucleus and CCF formation mentioned above. Despite these obvious differences, these cytoplasmic DNA types related to DNA damage have been confirmed to participate in the activation of cGAS-STING pathway. However, in aging T cells, cytoplasmic DNA can be sensed by KU complex (independent of cGAS-STING pathway), and they may also play an important role in physiology and diseases.
mitochondrial dna,mtdna
DNA(mitochondrial DNA, mtDNA) has thousands of copies in each cell and is densely packed into nucleoid. These nucleoids consist of a copy of mtDNA and various protein, such as mitochondrial transcription factor A (TFAM, a protein responsible for nucleoid structure, abundance and separation). Mitochondrial DNA is usually located in the matrix of mitochondria and encodes 37 genes: 13 kinds of mRNA (translated into some subunits of oxidative phosphorylation system), 2 kinds of ribosomal RNA and 22 kinds of transport RNA.
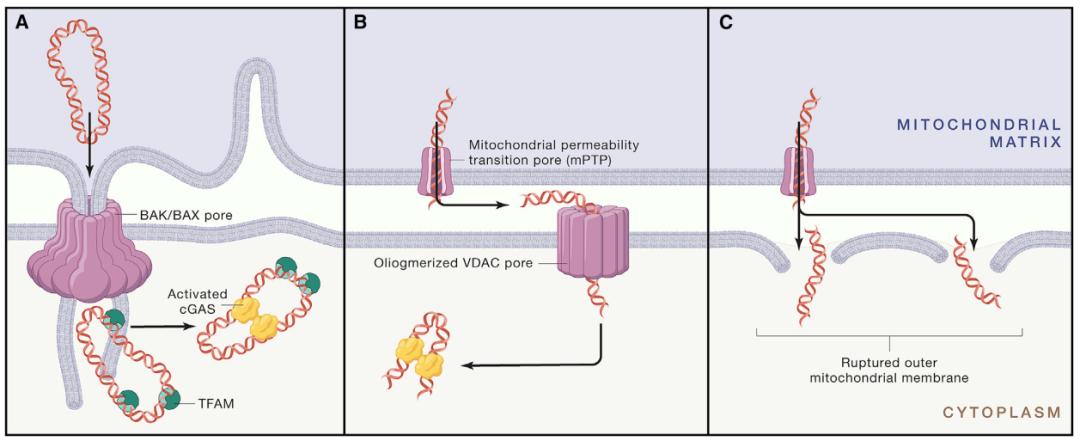
Fig. 3 release mechanism of mitochondrial DNA
(a–c) The release of mitochondrial DNA occurs through the following channels: (A)BAK and BAX channels provide the possibility for the inner membrane of mitochondria to pass through the outer membrane to form a protruding structure, and further allow matrix components such as mitochondrial DNA to escape; (b) mPTP channels in the inner membrane of mitochondria and oligomeric VDAC pores in the outer membrane of mitochondria; or (c) the outer membrane of mitochondria is broken due to the expansion of mitochondria mediated by mPTP. Once released, mitochondrial DNA can be sensed by DNA receptors. TFAM can enhance cytoplasmic cGAS induction.
Mechanism of mtDNA cytoplasmic release
Mitochondrial DNA in cytoplasm is related to the process of cell apoptosis. After receiving the apoptosis signal, pro-apoptosis proteins BAX and BAK are activated, and large holes are formed in the outer membrane of mitochondria, which leads to the increase of mitochondrial outer-membrane permeability (MOMP) (Figure 3). After the appearance of MOMP, the pores of BAX and BAK gradually widened, allowing the inner membrane of mitochondria to squeeze into cytoplasm. Then the prominent intimal permeability increased, which promoted the release of mitochondrial DNA. At first, MOMP was considered as an all-or-nothing event. All mitochondria in the cell showed MOMP, and pro-apoptotic proteins (such as cytochrome C) were released from the membrane gap, which eventually led to cell death. However, studies have shown that under sublethal stress, MOMP will occur in a small number of mitochondria, but it will not induce cell death. This phenomenon is called MOMP(minority MOMP, miMOMP). Therefore, mitochondrial DNA can also exist in the cytoplasm of cells that do not undergo apoptosis. Other mitochondrial stresses may also lead to cytoplasmic mitochondrial DNA leakage. For example, the decrease of TFAM expression leads to abnormal packaging, recruitment and distribution of mitochondrial DNA, which is finally released into cytoplasm.
Besides the lack of miMOMP and TFAM, another mechanism that may mediate the release of mitochondrial DNA into cytoplasm is the opening of mitochondrial permeability transition pore (MPTP) (Figure 3). MPTP is a transmembrane protein located in the inner membrane of mitochondria, which is usually opened by calcium accumulation in mitochondrial matrix, oxidative stress and other stressors. Although it has been proved that mPTP does not play a role in mitochondrial DNA leakage during apoptosis, a study shows that mitochondria will release mitochondrial DNA fragments by briefly opening mPTP in irradiated mouse brains. Another study shows that inducing oxidative stress in mitochondria isolated from rat hepatocytes can lead to the release of mitochondrial DNA fragments, and this process is partly mediated by the opening of mPTP. The above research shows that only mitochondrial DNA fragments will be released through mPTP, which is consistent with the fact that mPTP only allows molecules smaller than 1.5 kDa (smaller than mitochondrial DNA nucleoids) to be transported. However, the structural opening of pores may also lead to the expansion of mitochondria, the destruction of intima and the release of mitochondrial DNA cytoplasm, but further research is needed to confirm this conclusion. Interestingly, a recent study proposed a voltage-dependent anion channel (VDAC).The role of oligomerization in mitochondrial DNA release into cytoplasm. Studies have shown that mouse fibroblasts lacking mitochondrial endonuclease γ have more mitochondrial DNA, while mitochondrial endonuclease γ shows higher oxidative stress, which can be compensated by knocking out VDAC 1 and 3.
The dynamic changes of mitochondrial regulatory network also promote the release of mitochondrial DNA to cytoplasm. Mitochondrial DNA in TFAM-deficient fibroblasts increased, and mitochondria were elongated and highly fused. Mitochondrial division is very important for ensuring correct nucleoid distribution and removing damaged mitochondrial DNA. Consistent with this, the author found that the increase of mitogen 1 (MFN1) in TFAM-deficient cells was related to the decrease of the expression of interferon-stimulated genes (ISGs), which indicated that the destruction of mitochondrial regulatory network also affected the expression of interferon gene induced by mitochondrial DNA. A recent study also showed that knocking out GTP enzyme MxB located in the inner membrane of mitochondria would lead to mitochondrial breakage, inner membrane breakage and increase of mitochondrial DNA in cytoplasm. This result can support the viewpoint that the imbalance of homeostasis in mitochondrial regulatory network is the reason for the release of mitochondrial DNA into cytoplasm.
Function of cytoplasmic mtDNA
Mitochondrial DNA and bacterial DNA have many common characteristics. For example, mitochondrial DNA is hypomethylated compared with nuclear DNA. The unmethylated CpG motif in bacterial DNA is an effective trigger for inflammation. Interestingly, the similarity between mitochondrial DNA and bacterial DNA at the structural level probably reflects the bacterial origin of mitochondrial DNA, and is even considered as the basis of the joint action in immune signals. Mitochondrial DNA is considered as an exogenous substance outside mitochondria, and may cause inflammatory reaction. For example, in TFAM-deficient cells, the existence of mtDNA in cytoplasm is related to the increased expression of ISGs and depends on cGAS-STING-IRF3 pathway (Figure 3). In addition, the accumulation of cytoplasmic mitochondrial DNA caused by mitochondrial endonuclease γ deficiency has also been proved to induce the expression of ISGs in mouse fibroblasts, which indicates that cytoplasmic mitochondrial DNA is a major trigger of inflammatory reaction.
Cytoplasmic mitochondrial DNA has also been proved to play a role after virus infection. For example, although cGAS is more specific to cytoplasmic DNA than RNA, RNA virus can also activate cGAS-STING pathway. A study showed that the infection of human cells with dengue virus (DENV-2) led to the increase of mitochondrial DNA in cytoplasm and the activation of cGAS, which explained the mechanism of RNA virus activating innate immune signals. Consistent with the role of mitochondrial DNA in initiating antiviral immune response, TFAM-deficient cells showed high expression of type I interferon and ISGs when infected with herpes simplex virus 1 (HSV-1), and it was more resistant to infection than wild-type cells. Treating TFAM-deficient fibroblasts with dideoxycytidine, ddC) can eliminate the antiviral initiation and resistance to virus infection. ddC can inhibit the replication of mitochondrial DNA in these cells and reduce the stress of mitochondrial DNA, which indicates that cytoplasmic mitochondrial DNA enhances the antiviral innate immunity. Interestingly, another recent study showed that microbial infection induced the cytoplasmic release of miMOMP and BAX/BAK-dependent mitochondrial DNA, which would lead to the activation of cGAS-STING and the secretion of pro-inflammatory cytokines.Blocking the signal of sublethal apoptosis can damage the ability of cells to resist pathogenic infection, which means that the release of mitochondrial DNA cytoplasm mediated by BAX/BAK is an effective immune defense mechanism.
In addition to triggering inflammation through cGAS-STING recognition, cytoplasmic mitochondrial DNA can also activate other receptors in cells, eventually leading to downstream inflammation. For example, mitochondrial DNA has been proved to activate NLRP3 inflammatory corpuscles. A study shows that treating cells with mitochondrial dysfunction with lipopolysaccharide or ATP will lead to the increase of mitochondrial DNA level in cytoplasm, which is helpful to secrete interleukin (IL)-1β and IL-18 through NLRP3 inflammatory corpuscles. The author also suggested that NLRP3 not only acts as a downstream receptor of cytoplasmic mitochondrial DNA, but also promotes the release of mitochondrial DNA by promoting the formation of mPTP in mitochondrial inner membrane. Another study showed that during the process of macrophage apoptosis, oxidized mitochondrial DNA was released into the cytoplasm and combined with NLRP3 inflammatory body to activate. In addition, a recent study also showed that macrophages triggered the new synthesis of oxidized mitochondrial DNA fragments and activated NLRP3 inflammatory bodies, which indicated that NLRP3 was more likely to bind to oxidized mitochondrial DNA.
Cytoplasmic mitochondrial DNA and disease treatment
It has been reported that mitochondrial DNA circulating in plasma is related to several specific age-related diseases. For example, studies have shown that the level of mitochondrial DNA in plasma and joint fluid of patients with rheumatoid arthritis is significantly increased. Rheumatoid arthritis is a disease characterized by chronic inflammation, with a high incidence among the elderly. The levels of pro-inflammatory factors tumor necrosis factor alpha (TNF-α) and mitochondrial DNA in blood circulation of patients with type 2 diabetes mellitus are increased, and the latter is related to insulin resistance. In addition, high levels of circulating mitochondrial DNA in plasma were found in patients with ovarian cancer, testicular cancer, prostate cancer and lung cancer. The concentration of mitochondrial DNA was also increased in plasma and bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis (IPF). Plasma mitochondrial DNA level is related to disease progression and has been proved to predict mortality. In addition, normal human lung fibroblasts treated with isolated mtDNA will express high levels of α -smooth muscle actin, which is a characteristic of fibrosis diseases, indicating that mitochondrial DNA plays a role in the pathophysiology of IPF.
Although it is not clear whether circulating mitochondrial DNA is related to age-related diseases, due to its powerful pro-inflammatory properties, cytoplasmic and extracellular mitochondrial DNA may promote the development of diseases to some extent. Therefore, mitochondrial DNA and its release mechanism may be potential therapeutic targets for improving or preventing diseases related to the elderly. For example, studies have shown that the expression of Bax in alveolar epithelial cells of IPF patients is increased, which indicates that MOMP is involved in the occurrence and development of the disease. Consistent with this, the treatment of IPF mouse model with BAX-inhibiting peptide V5 (BIP-V5) can inhibit the activation of BAX in cytosol, reduce inflammation, improve lung pathology and improve the survival rate of experimental mice. In addition, the deletion of Bax gene has been proved to improve the ovarian function of elderly female mice, and promote the significant improvement of many age-related phenotypes, such as increasing bone density and intensity, better maintaining weight and reducing anxiety. Another study showed that the inhibition of BAX by two small molecule BAX channel inhibitors, Bci1 and Bci2, had protective effects during cerebral ischemia in gerbils. After reperfusion, the administration of BAX inhibitor can effectively inhibit MOMP, which shows that the release of cytochrome C in neuronal cytoplasm is reduced and the hippocampal damage is significantly reduced. These studies show that inhibiting MOMP by targeting BAX may be a valuable therapeutic strategy, which can improve the phenotype of age-related decline and prolong healthy life. Besides,In the mouse model of systemic lupus erythematosus, inhibiting VDAC oligomerization by using inhibitor VBIT-4 has been proved to reduce mitochondrial DNA release and IFN signal transduction, and improve the severity of the disease, which indicates that inhibiting VDAC oligomerization and mPTP may be an effective therapeutic strategy and can improve related diseases involving mitochondrial DNA release in cytoplasm.
All kinds of SASP play a role in paracrine way, inducing the aging of surrounding cells, which is considered as a mechanism of aging-related tissue dysfunction caused by aging cells. IL-1β is a major SASP factor, which can induce the release of mitochondrial DNA and then activate pro-inflammatory reaction. This may be one of the mechanisms by which aging cells stimulate the release of mitochondrial DNA in the cytoplasm of neighboring cells, thus aggravating inflammation, but further research is needed to confirm this hypothesis. A recent study shows that aging cells are the main source of extracellular mitochondrial DNA accumulated with aging. The pharmacological action of removing aging cells from aged animals significantly reduced circulating mitochondrial DNA, improved age-related inflammation and prolonged the survival time of mice receiving organ transplantation from aged animals. Although the mechanism of mitochondrial DNA release into extracellular space has not been fully clarified, this study shows that cell aging is an important factor for the increase of circulating mitochondrial DNA with age. In addition, damaged mitochondria in aging cells may contribute to the release of mitochondrial DNA and promote chronic inflammation related to aging, which is a subject worthy of further study. Eliminating aging cells has been proved to be an effective strategy to improve many age-related diseases. Although the role of mitochondrial DNA in this respect remains to be determined, targeting aging cells or simultaneously targeting the release of mitochondrial DNA from aging cells is another intervention strategy worth trying.
Retrotransposon
Transposable elements, TEs) is a mobile DNA sequence discovered by Barbara McClintock in 1950. Transposons account for 50% of the human genome, and contribute significantly to genetic variation at the population level. There are two types of transposons: one is a class II transposon called DNA transposon, which codes for protein transposase, allowing these transposons to mediate insertion and excision functions in the genome through the "cut and paste" mechanism. The other is class I transposons or retrotransposons, which rely on RNA mediators to transfer their transposons to other parts of the genome because they do not encode transposase. Retrotransposons are the main transposons in human genome, which can be divided into two types: LTR retrotransposons, including long terminal repeat at both ends, and non-LTR which does not contain these repetitive sequences. Non-LTR can be subdivided into long scattered repetitive sequences (LINEs) and short scattered repetitive sequences (SINEs). SINEs includes Alu family sequence, which is about 300 bp in length, and has more than one million copies in the genome, accounting for 11% of the human genome. In contrast, the length of LINE-1(L1) family sequence can reach 6 KB, and the number of copies exceeds 500,000, accounting for 17% of human genetic material. It is the most abundant human LINE. In addition, the L1 family includes the only endogenous autonomous reverse transcription factor that still retains the ability of autonomous reverse transcription transposition.Its protein products (ORF2p and ORF1p) are also responsible for mediating the reverse transcription transposition of sinusoidal involuntary elements.
Activation of retrotransposon and formation of cytoplasmic retrotransposon cDNA
After activation, LINEs are transcribed by RNA polymerase II, and the resulting mRNA is transcribed into cytoplasm, where it is translated into two kinds of protein: ORF1p (an RNA binding protein) and ORF2p as an endonuclease and a reverse transcriptase (Figure 4). These two proteins form a complex with mRNA transcripts and then relocate back to the nucleus. In the nucleus, the reverse transcription transpositional time occurs by target-primed reverse transcription (TPRT) when the target is not determined. In the process of TPRT, some people think that the endonuclease encoded by L1 cuts the negative sense chain of the target DNA and exposes a 3′-terminal hydroxyl group, which can be used as a primer for ORF2p to reverse transcribe L1 mRNA. After the synthesis of the second strand DNA, the mechanism of the reinsertion process has not been clarified. Although TPRT occurs in the nucleus, under some conditions, L1 cDNA is also found in the cytoplasm, such as autoimmune reaction. Here, we discuss the possible mechanisms that two kinds of L1 cDNA exist in cytoplasm: (1) The accumulation of a large number of L1 mRNA and L1 protein in cytoplasm may lead to reverse transcription without standard DNA template, thus leading to the increase of cytoplasmic cDNA elements; (2) In the process of TPRT, the generated cDNA will not be reintegrated into the genome, but will leave the nucleus and enter the cytoplasm through an unknown process.
DNA damage is related to the activation of retrotransposon. Studies have shown that the exposure of mouse and human cells to DNA damaging agents, such as etoposide, ultraviolet rays and γ-radiation, will lead to an increase in the expression level of AlRNA and an increase in reverse transcription translocation. In fact, after X-ray irradiation, new insertion of SINE and LINE-1 was detected in mouse germ cells. Although the mechanism of DNA damage promoting reverse transcription transposition is not clear, it may be that the change of transcription factor expression after DNA damage induction promotes the transcription of these elements. Epigenetic changes caused by some genotoxic stresses may also provide another mechanism of reverse transcription transposition induced by DNA damage. For example, methylation of CpG site at 5’UTR site will hinder the activity of L1 promoter and prevent reverse transcription transposition. The finding that MeCP2 can inhibit L1 expression and reverse transcription transposition also supports this view. Studies have shown that oxidative damage reduces the affinity of MeCP2 with damaged methylated DNA. Therefore, the oxidative damage near L1 element may show inhibition. Since the accumulation of DNA damage is one of the characteristics of cell aging and individual aging, the increase of DNA damage may contribute to the reactivation of retrotransposons observed in aging cells and aging tissues.
More and more evidences show that extensive epigenetic changes during cell aging and individual aging explain the reactivation of retrotransposons. In fact, a previous study showed that the increase in L1 transcription observed during cell aging was mediated by the decrease in the expression of RB1 and the increase in the expression of transcription activating factor FOXA1, in which RB1 is a transcription inhibitory factor that promotes heterochromatization of L1 elements. Another mechanism of age-related retrotransposon activation is the decrease of SIRT6 expression, which is a histone deacetylase and has been proved to regulate life span. SIRT6 inhibits L1 activity by binding to the 5’UTR of L1 element, and promotes heterochromatization by promoting the interaction between KAP1 and heterochromatin factor HP1α, thus making L1 element lose its transcriptional activity. However, in the process of cell aging and in the brain of aged mice, SIRT6 at L1 locus is deleted, which allows L1 expression and reverse transcription transpositions to occur. Consistent with this, SIRT6-deficient mice showed an accelerated aging phenotype, and the expression of L1 increased in many tissues. SIRT7, another member of sirtuin family, is also responsible for the epigenetic regulation of LINE1. LINE-s tends to locate in the protein-related domains of the nuclear laminae, which are mainly located in the periphery of the nucleus. Importantly, SIRT7 mediates the deacetylation of H3K18 in L1 element, thus promoting its association with nuclear laminin.Keep it in a transcriptional active state. One possibility is that in the aging process, due to the lack of Lamin A/C, SIRT7 can’t fix L1 element on the nuclear fiber layer, thus starting the expression of LINE-1 and reverse transcription transpositions.
Reversing the function of transposon
The inhibition of reverse transcription transposition will be relieved in some physiological processes. For example, during preimplantation development, when methylation modification is absent, the expression of retrotransposon is relatively high in late oocytes and early embryos of mice, and decreases in later stages. Evidence of L1 reverse transcription transposition was also reported during human early embryogenesis. In addition, reverse transcription transposition may also occur in the process of neuronal differentiation and affect the fate of cells. In addition, many stressors can induce the expression and translocation of retrotransposons, such as genotoxic stress, heat shock, viral infection and heavy metals. Retrotransposon activation can also be found in aging cells. In aging cells, the chromatin structure of many retrotransposon families becomes looser, which increases their transcription and eventually leads to the occurrence of retrotransposon of these elements, such as Alu, SVA and L1. Nevertheless, it is not clear whether the activation of retrotransposons is beneficial. However, the programmed activation of retrotransposons in aging cells and the subsequent activation of cGAS-STING-dependent IFN-I response and pro-inflammatory phenotype are beneficial to tumor inhibition related to cell aging.
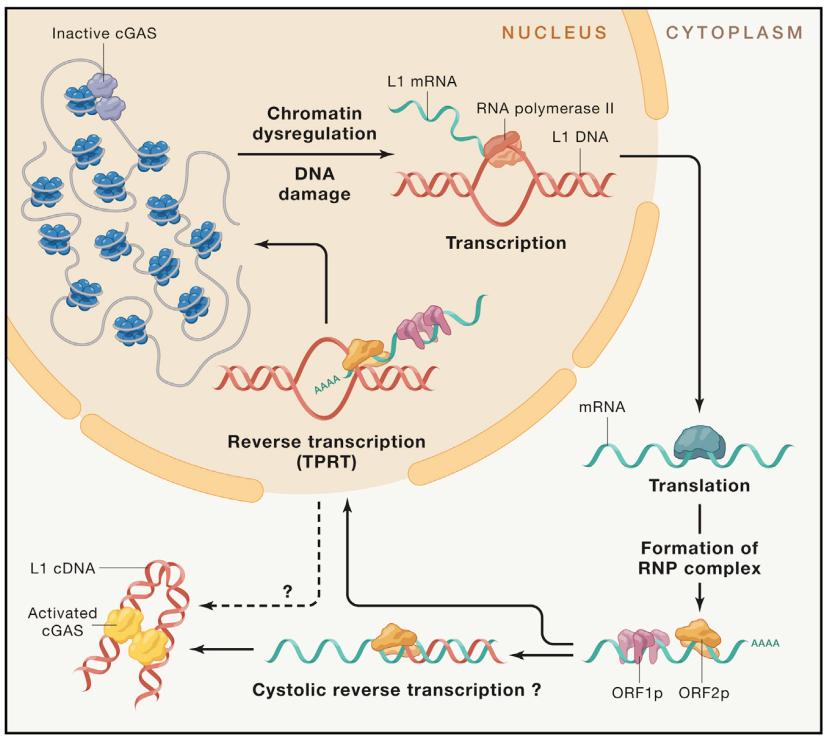
Fig. 4 Mechanism of cytoplasmic DNA production caused by retrotransposon activation.
After transcriptional activation, polyadenylation (polyA) LINE-1 mRNA will encode ORF1p and ORF2p, which will be transported to the cytoplasm for translation. ORF1p and ORF2p combine with mRNA to form a ribonucleoprotein (RNP) complex. Reverse transcription and L1 nuclear integration are completed by TPRT mechanism which depends on the annealing reaction of thymidine nucleotide poly(A) tail in nuclear DNA. In addition, reverse transcription may also occur in cytoplasm. L1 cDNA accumulates in cytoplasm during aging and diseases, and is detected by nucleic acid sensor.
Retrotransposon dysfunction
The unrestricted movement of L1s in genome may be harmful, and more than 60 human genetic diseases are attributed to the insertion of retrotransposons. Therefore, the transcription of retrotransposons is strictly regulated and silenced at many levels, including epigenetic modification, premature termination of transcription, miRNAs(micro-RNAs), siRNAs(small-interfering RNAs) and post-transcriptional modification.
Some studies provide new evidence that the increased expression of retrotransposon gene is related to aging and diseases. For example, the expression of different retrotransposons and retrotransposable events in Caenorhabditis elegans, yeast, Drosophila melanogaster, mice and human cells increased with age. In the aging model of Saccharomyces cerevisiae, with the increase of age, the mobility of Ty1 in the yeast population increases, and this reverse transcription transposition is related to chromosome rearrangement. In addition, the expression of Cer1 retrotransposon in Caenorhabditis elegans increases with age, while in Drosophila, the increase of age-related gypsy and non-LTR retrotransposons R1 and R2 is related to the loss of neuronal function and the decline of cognitive ability. In mammals, the expression of retrotransposon is increased in the liver and skeletal muscle of aged mice, which leads to the occurrence of retrotransposon in elderly individuals. Interestingly, these phenomena can be weakened by calorie restriction, which has been proved to prolong the life span of a large number of organisms and reduce the burden on aging cells. Retrotransposon activation is also observed in mammalian cells lacking typical aging markers and aging model organisms that do not show cell aging, which indicates that retrotransposon activation is an inherent and conservative feature of aging process.
The activation of retrotransposons has also been proved to promote age-related chronic inflammation. In the later stage of senescence, the reactivation of LINE-1 element is accompanied by the accumulation of L1 cytoplasmic cDNA, which is partly due to the decrease of exonuclease’s TREX1 level and the activation of cGAS-STING-dependent IFN-I response and pro-inflammatory phenotype. Consistent with this, SIRT6 knockout mouse fibroblasts showed L1 activation, L1 cDNA accumulation in cytoplasm and increased expression of type I IFN gene. Immunocoprecipitation analysis of these cells showed that the abundance of L1 DNA bound to cGAS increased, which confirmed that L1 DNA in cytoplasm bound to cGAS and activated IFN-I reaction. Using nucleoside reverse transcriptase inhibitor (NRTIs) to inhibit the synthesis of L1 DNA in cytoplasm or knock down the expression of L1 by shRNA can reduce the activation of IFN gene and the level of SASP, which indicates that the accumulation of L1 cDNA in the cytoplasm of aging cells is an important factor to maintain the pro-inflammatory phenotype. Interestingly, increased expression of cytoplasmic L1 DNA and IFN genes has been observed in the tissues of many aged mice.
Transcription intermediates independent of reverse transcription and derived from retrotransposons are also related to the development of age-related diseases. It has been shown that in an age-related macular degeneration called geographic atrophy, GA), the decrease of the miRNA processing enzyme DICER1 in retinal pigment epithelium (RPE) cells leads to the accumulation of AlRNA. The latter then activates the inflammatory corpuscles of NLRP3, stimulates the production of cytokines and the cytotoxicity of RPE cells induced by MyD88, which leads to map atrophy. In addition, the study found that L1-derived mRNA was highly expressed in synovial fluid of patients with rheumatoid arthritis, suggesting that L1 activity may play a role in the invasive phenotype of rheumatoid arthritis.
Study on targeted therapy of retrotransposon
The most widely studied treatment to inhibit L1 reverse transcription transposition is to use nucleoside analogue reverse transcriptase inhibitor (NRTIs), which is a kind of antiviral compound that inhibits nucleic acid polymerase, including reverse transcriptase, and is also used to treat AIDS. A study of LINE-1 retrotranspositional test in vitro shows that L1 retrotranspositional can be inhibited by many reverse transcriptase inhibitors with different effects. Consistent with this, another group of studies showed that reverse transcriptase inhibitors can effectively inhibit the reverse transcriptase activity of L1 protein ORF2p, thus reducing reverse transcription transpositions. In addition, NRTIs has been proved to eliminate the accumulation of cytoplasmic L1 DNA in aging cells. Based on the IFN-I reaction triggered by cytoplasmic L1 DNA, which leads to age-related inflammation, NRTIs can be used as a potential treatment for age-related diseases. In fact, the treatment of aged mice with NRTIs effectively reduced L1 cytoplasmic DNA and subsequent inflammation in various tissues, and improved some age-related phenotypes, such as tissue macrophage infiltration, renal glomerulosclerosis and skeletal muscle atrophy. A similar improvement in healthy life expectancy was observed in SIRT6 knockout progeria-like mice treated with NRTIs. In addition, the increase of LINE-1 reverse transcription transposition is related to the decrease of oocytes in fetal period, which will lead to more than two-thirds of oocytes dying before birth. Studies have shown that the treatment of pregnant female mice with nucleoside analogue AZT can improve oocyte loss,Improve the vitality of oocytes in embryos. This raises the possibility that reverse transcriptase inhibitors may prolong the reproductive life of women by targeting L1 element.
Although mouse studies show that NRTI is an effective therapeutic strategy for L1-driven age-related inflammation and pathology, long-term NRTI therapy has been proved to induce adverse side effects in human patients, such as hepatotoxicity, because it can also inhibit mitochondrial DNA polymerase γ. In addition, NRTIs can also inhibit telomerase, which is considered to contribute to the accelerated aging phenotype associated with HIV patients. Therefore, it is necessary to formulate more specific intervention measures to safely and effectively treat diseases caused by uncontrolled L1 activities. For example, promoting the degradation of cytoplasmic L1 DNA to reduce inflammation may be a promising direction of targeted therapy.
Prospect and conclusion
Since 1960s, it has been known that exogenous nucleic acids can stimulate immune response. However, this pathogen induction strategy also allows the detection of endogenous nucleic acids that are wrongly positioned or abnormal. In recent years, breakthroughs in biomedical research fields such as cancer, immunology, aging, DNA damage and chromatin regulation have accelerated the research progress on the molecular mechanism of endogenous cytoplasmic DNA formation and function. Although it is known that the cytoplasmic localization of endogenous DNA triggers the regulation of signal pathways related to many chronic diseases, there are still many unknown information to be discovered. The author puts forward two main questions in this paper: (1) What is the mechanism of the formation and function of endogenous cytoplasmic DNA? (2) How does the interaction between endogenous cytoplasmic DNA pathways maintain normal physiological functions and drive the occurrence and development of diseases? Further understanding of the similarities, differences and interactions between heterogeneous DNA and related pathways will provide therapeutic opportunities for the treatment of human diseases.
Common Mechanism of Cytoplasmic DNA Formation and Function
There may be a common mechanism between the formation and function of heteroplasmic DNA, and a summary of these common mechanisms can produce a new insight. Firstly, by comparing the formation and signal transduction mechanism of endogenous cytoplasmic DNA, we can deeply understand the role of cytoplasmic DNA in physiology and disease, and reveal many similarities. For example, the loss of nuclear membrane integrity in aging cells and fibrotic diseases, such as HGPS, are consistent with the loss of nuclear membrane integrity in MN. These similarities include abnormal nuclear membrane function, changes in nuclear fiber layer structure, accumulation of DNA damage and activation of downstream cGAS. The loss of nuclear membrane integrity is also a potential mechanism of cytoplasmic localization of retrotransposons, although the cytoplasmic reverse transcription of L1 may also be one of the mechanisms. Second, although the relevant mechanism is not clear, CCF and mitochondrial DNA are considered to be partially localized in endosomes, and then they are induced, degraded or possibly secreted by cells. The degradation of dnase, such as the treatment of all forms of endogenous cytoplasmic DNA by lysosomal DNase2 or cytoplasmic TREX, can be used as a negative feedback pathway to inhibit the activation of DNA sensors. CCF and MN are also thought to be degradable by autophagy. Recent studies have shown that cGAS itself can be used as an autophagy ligand for MN degradation, which indicates that autophagy degradation may be the common fate of cytoplasmic DNA related to cGAS. Various forms of cytoplasmic DNA may be secreted and used as involuntary signals of cells in physiology and diseases, which is considered as a biomarker of aseptic inflammation.Thirdly, different cytoplasmic DNAs have similar signal pathways, most notably cGAS-STING pathway, although mitochondrial DNA, MN and other DNA fragments related to DNA damage are also thought to activate AIM2 and NLRP3 inflammatory body pathways. It will be interesting to verify whether additional DNA sensors are also shared between cytoplasmic DNA, and the functional mechanism of these sharing is consistent with the evolutionary conservative strategy of monitoring foreign DNA as a danger signal. Interestingly, many species of bats have evolved to naturally tolerate cytoplasmic DNA by reducing their perception of it. Fourthly, many kinds of cytoplasmic DNA observed in aging cells correspond to the role of foreign DNA as an activator of cellular antiviral mechanism, which strengthens the connection between cell aging and antiviral programs. Specifically, cell senescence can be induced by virus infection, and aging cells can activate antiviral pathways, and the replication of many viruses may be damaged by SASP of aging cells. Notably, in COVID-19 animal model, senolytic therapy eliminated virus-induced aging cells and reduced inflammation and mortality, which indicated that aging cells were potential therapeutic targets for virus infection. At the molecular level, the key regulatory factors of aging cell phenotype are also the core of virus defense mechanism, including tumor suppressor pRB and p53, histone chaperone HIRA, PML nucleosome, and cGAS, STING and ISGs.It is found that the additional shared and conservative "seno-viral" mechanism may provide information for the therapeutic strategy of targeting multiple cytoplasmic DNA at the same time, such as regulating SASP in aging cells, which may be driven or enhanced by CCF, mitochondrial DNA and retrotransposon.
Different mechanisms of formation and function of cytoplasmic DNA
Although there are basic similarities between endogenous cytoplasmic DNA, detailed research reveals their mechanism differences and many unresolved problems (Figure 5). As for the formation mechanism, the time characteristics of cytoplasmic localization of CCF, mitochondrial DNA and retrotransposon and the dynamic changes of cytoplasmic DNA induction in time are still unclear. For example, in aging cells, the formation of CCF occurs after cell cycle arrest, which is consistent with the independent formation mechanism of cell cycle. However, the direct role of MN and mitochondrial DNA in this process is still unclear. Aging cells also show retrotransposon activation, however, this activation only occurs in the late senescence, which is different from CCF formation in the late senescence days. Because these processes may occur in the same cell, which are related to similar formation mechanisms (such as nuclear membrane dysfunction and DNA damage) and activate cGAS, it will be very interesting to analyze this time relationship. The authors speculate that the formation of CCF in aging cells degrades unrepaired DNA, changes the epigenome structure in a gradual way, and finally suppresses retrotransposons. The temporal evolution of senescence cell phenotype has been observed in the process of "deep senescence" before.
The regionalization of cytoplasmic DNA in cytoplasmic chamber is not clear. Electron microscope showed that the aging cell CCF induced by oncogene could be located in autophagosomes with double membranes, and fluorescence microscope showed that CCF was co-located with lysosomes and activated by giant phagocytosis. However, it is not completely clear whether CCF is always consistent with the formation of autophagy bodies, or whether autophagy degradation is the only fate of CCF. Similarly, mitochondrial DNA is known to locate in endosomes and activate DNA receptors and pro-inflammatory mediators TLR9. Because the endosome is topologically adjacent to the outside of the cell, the location of DNA endosome is usually considered to depend on the endocytosis of extracellular DNA. This raises the question of how cytoplasmic mitochondrial DNA enters the endosome. The localization of CCF in autophagosome shows that the transport between autophagosome and other endocytosis compartments is another potential localization mechanism. Consistent with this hypothesis, antimicrobial peptide LL37 has been proved to promote the transport of aggregated DNA structures to early endosomes, promote the activation of TLR9 and the production of interferon. Further study on cytoplasmic DNA transport in cytoplasm can provide information for therapeutic strategies to degrade cytoplasmic DNA or prevent its secretion. Both of these methods have therapeutic potential for autoimmune diseases.
Although most of the research focuses on cGAS as the main sensor of cytoplasmic DNA, the regulation of cGAS seems to be upstream and downstream related. For example, although the activation of MN-dependent cGAS interferon gene has been well studied in cancer cells, the activation of cGAS in aging cells is usually related to NF-κb-dependent cytokine response, which is considered to be partially dependent on the activation of p38-MAPK in aging cells. In addition, there are some other potential factors that will affect the cGAS pathway. Recently, the research on cGAS’ induction of DNA has gradually revealed a complex regulatory network, including phosphorylation inhibition, nucleosome and chromatin structure binding inhibition, manganese activation, DNA damage-induced aging coactivator activation, and TFAM activation in mitochondrial DNA perception. At least in some cases, the localization of endogenous cGAS is considered to be in the nucleus and combined with nucleosome in an inactive state, but the exact mechanism of cGAS activation (such as MN disruption) is still unclear. Recently, there is evidence that cytoplasmic transport of nuclear cGAS is necessary for cytoplasmic DNA perception, at least in the context of exogenous DNA transfection and vaccinia virus infection. However, compared with being recruited from cytoplasm, the signal transduction function of cGAS transported from the main nucleus to MN and CCF needs to be further explored. Given that protein -DNA complex plays a central role in DNA function, it is not surprising that the cytoplasmic localization of DNA is regulated by specific environment and cytoplasmic protein components. of course,It is reported that cGAS can sense all major cytoplasmic DNA, but targeting other DNA sensors to detect cytoplasmic DNA, specific protein of cytoplasmic DNA source and specific protein of cytoplasmic subcellular can also play an important role in function and therapeutic targeting. In fact, the protein omics characteristics of CCF have identified these targets. Although the enrichment of heterochromatin-related histone markers indicates that CCF is produced by heterochromatin, the genomic region or specific site that produces CCF is not clear. Single cell genome sequencing has greatly improved the understanding of the mechanism of MN formation and chromosome breakage, which shows that similar methods can answer this outstanding question in CCF biology, and determining the DNA sequence of CCF can provide information for the targeting and formation mechanism of treatment.
Finally, although cGAS-STING pathway inhibitors are promising strategies for treating chronic inflammation and some cancers, the use of these drugs must take into account the diversity of endogenous cytoplasmic DNA functions. For example, the cGAS-STING signal that inhibits chronic inflammation downstream of CCF activation may be beneficial with age, but this strategy may also damage the signal transduction of intracellular immune monitoring and tumor inhibition initiated by MN. However, CCF and MN are difficult to distinguish under the microscope at present, and are often confused (Table 1). Therefore, the treatment methods must consider the specific endogenous cytoplasmic DNA, including the formation mechanism, localization and dynamics, as well as the interaction between different cytoplasmic DNA in the presence of multiple types, such as aging cells.
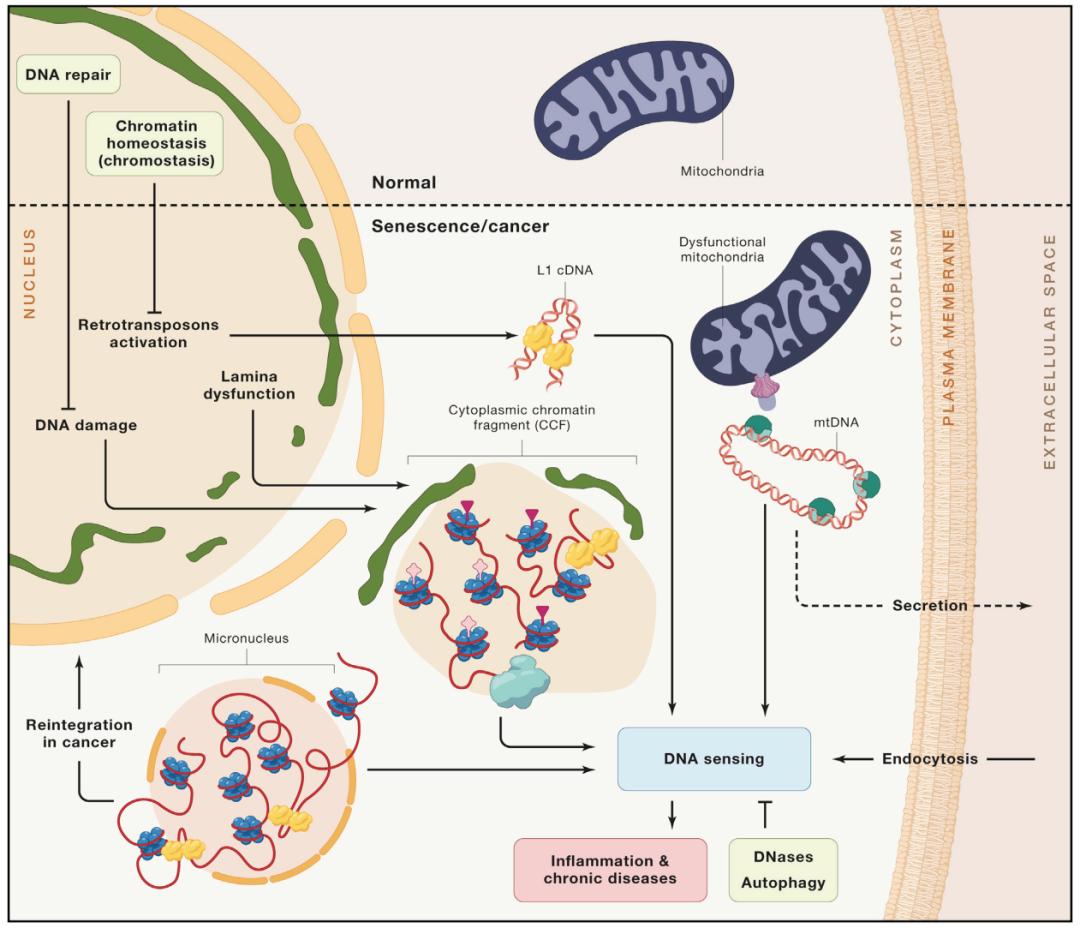
Fig. 5 types of cytoplasmic DNA and their perception of aging and diseases
Endogenous cytoplasmic DNA is produced by different mechanisms, but it is sensed and processed by similar cellular pathways. These mechanisms ensure chromatin homeostasis and DNA repair, as well as the catabolism of cytoplasmic DNA, and prevent the overactivation of innate immune signals. The loss of these homeostatic processes is considered to be one of the causes of inflammation and chronic diseases.
Conclusion
Whether exogenous or endogenous, DNA in cytoplasm can be used as a powerful stimulus signal to activate innate immune response. Although this field has long been concerned about the pathogenic activation of these pathways, recent work has found that endogenous cytoplasmic DNA is the main trigger of chronic diseases, especially cancer. These chronic diseases (including cardiovascular diseases, neurodegenerative diseases, sarcopenia, etc.) have common typical characteristics-patients are all elderly people. Chronic inflammation is partly caused by cytoplasmic DNA, which is related to many mechanisms and pathological changes reported in aging biology, which indicates that cytoplasmic DNA is one of the potential therapeutic targets to prolong human healthy life. Although preclinical studies on cytoplasmic DNA show that it is expected to be a target for the treatment of human health and aging-related diseases, there are still many challenges in its clinical application. Understanding the mechanism of cytoplasmic DNA formation and signal transduction, especially the subtype-specific mechanism of cytoplasmic DNA formation and sensing, will help to realize the treatment of clinical diseases, especially chronic diseases.
Original title: Cytoplasmic DNA and Aging and Diseases
Read the original text